Research
Generative AI & Molecular Design
AI/MLDrug Discovery
A drug candidate must be potent, selective, safe, metabolically stable, and possible to synthesize. Optimizing all of these properties at once is the central challenge of molecular design. I develop generative models that navigate this landscape through two complementary modes. De novo generation explores broad chemical space to discover novel molecular scaffolds. Series-constrained generation modifies specific positions on a known active molecule, exploiting accumulated structure-activity relationships during lead optimization. Synthesizability is enforced as a hard constraint: every generated candidate is filtered against known reaction templates and commercially available building blocks, so compounds are synthetically accessible.
Molecular Property Prediction
AI/MLCheminformatics
I developed stacked ensemble models that combine graph neural network representations with molecular fingerprints and physicochemical descriptors to predict absorption, distribution, metabolism, excretion, and toxicity (ADMET) endpoints. Curriculum learning substantially improved generalization. Training first on large public datasets, then fine-tuning on the target distribution, helped the models remain robust across structurally diverse compounds. Cross-validation with both chemical-similarity and temporal splits produced generalization estimates resilient to distribution shift. Evaluated on fully held-out compound libraries in the OpenADMET-ExpansionRx Blind Challenge, this approach placed 18th out of 400+ submissions across 9 ADMET endpoints.
Leaderboard: OpenADMET-ExpansionRx Challenge · Code: OpenADMET-ExpansionRx-Blind-Challenge
Polyelectrolyte Complexation & Ion Binding
PhysicsSimulation
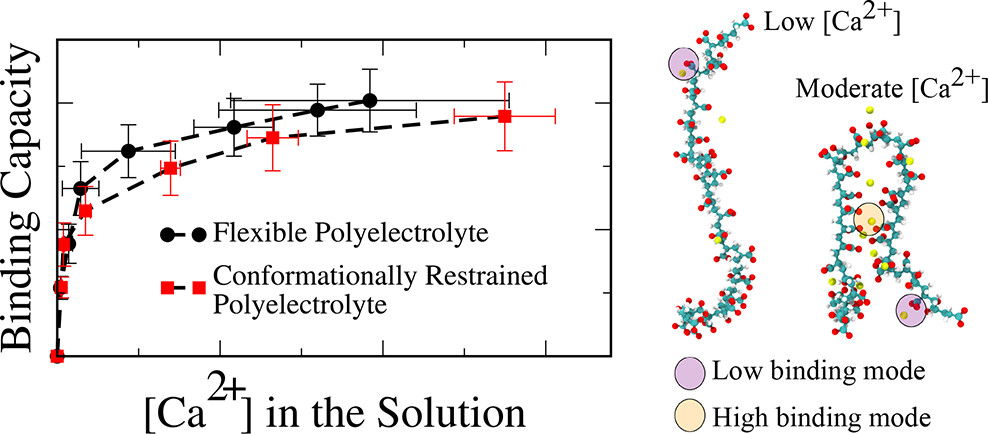
Charged polymers (polyelectrolytes) in water bind multivalent ions like Ca²⁺, a property exploited in water softening, drug delivery, and scale inhibition. Yet the molecular mechanisms of this binding were not well understood. Using well-tempered metadynamics and Hamiltonian replica exchange, I calculated binding isotherms and free-energy landscapes for Ca²⁺ on poly(acrylic acid), identifying two dominant binding geometries distinguished by carboxylate coordination number. All-atom MD combined with unsupervised deep learning mapped concentration-dependent phase diagrams. Surface adsorption studies showed that water-mediated hydrogen bonds, not direct polymer-surface contacts, govern binding on CaCO₃. This work was conducted in collaboration with Dow Chemical.
Key papers: Langmuir 2025 · Macromolecules 2024 · Langmuir 2024
AI/ML Methods in Soft Matter
AI/MLDeep Learning
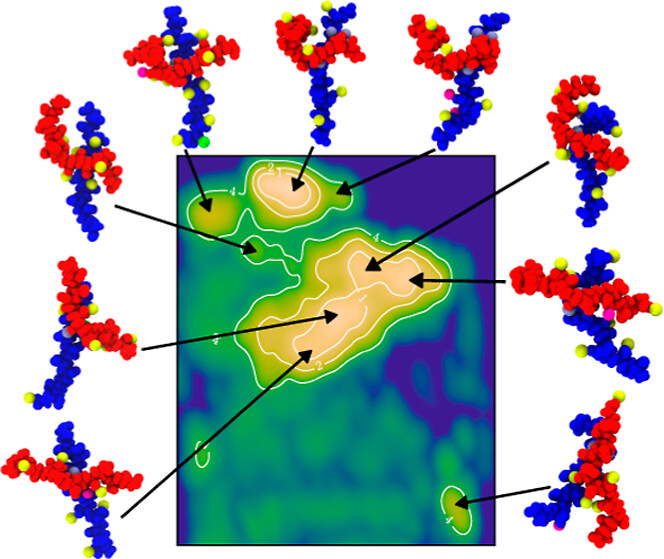
Polyelectrolyte simulations produce high-dimensional free-energy landscapes where interpretable structure-property relationships are hard to extract directly. I developed workflows coupling enhanced-sampling molecular dynamics with dimensionality reduction and clustering, identifying dominant polymer conformations and their thermodynamic stability from trajectory data. I also trained denoising diffusion probabilistic models (DDPMs) to generate polymer conformational ensembles consistent with Boltzmann statistics. These models offer a fast generative complement to physics-based sampling for polymer design.
Code: DDPM-Enhanced-Sampling
Microhydrodynamics & Active Matter
PhysicsFluid Dynamics
Schools of fish, flocks of birds, and bacterial colonies all display spontaneous collective ordering. I set out to understand how much of this behavior emerges from fluid mechanics alone, without phenomenological interaction rules. I derived an analytical framework for self-propulsion in potential flow showing that a deformable body achieves net displacement by exploiting its configuration-dependent added mass, without performing net work on the fluid. Self-propulsion had previously been thought to require viscous dissipation, which is absent in potential flow. I also developed C++/CUDA boundary integral simulations for many-body Stokes flow, demonstrating that purely hydrodynamic coupling is sufficient to produce collective ordering in active particle suspensions.
Key paper: J. Fluid Mechanics 2022

Lipid Membrane Mechanics
PhysicsTheory
Lipid membranes are unusual materials. Lipids flow in-plane as a two-dimensional viscous fluid while the membrane bends out-of-plane as an elastic shell. The coupling between these two behaviors had been largely overlooked. Using differential geometry and linear irreversible thermodynamics, I developed a continuum theory for membrane dynamics across planar, spherical, and cylindrical geometries. This work introduced the Scriven-Love number, a dimensionless ratio quantifying when intramembrane viscous stresses matter relative to elastic bending forces. Evaluating this number across physiological processes showed that in-plane viscosity cannot generally be neglected.
Key paper: Physical Review E 2020